人类泛基因组参考将使人们能够更完整和公平地了解基因组多样性
加州大学圣克鲁斯分校的科学家和一个研究人员联盟发布了第一个人类泛基因组的草案 - 一种新的,可用的基因组学参考,结合了来自不同祖先背景的47个个体的遗传物质,以便更深入,更准确地了解全球基因组多样性。
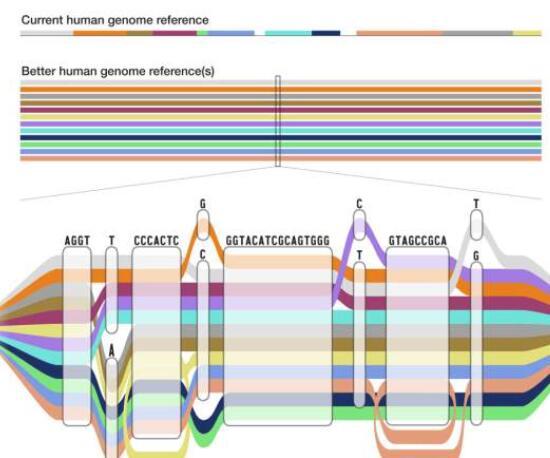
通过将119.<>亿个碱基(DNA序列中的“字母”)添加到现有的基因组学参考中,泛基因组提供了人类遗传多样性的表示,这是单个参考基因组无法实现的。它非常准确,更完整,并显着提高了对人类基因组变异的检测,正如今天发表在《自然》、《基因组研究》、《自然生物技术》和《自然方法》杂志上的一系列开创性论文所示。
该泛基因组由人类泛基因组参考联盟(HPRC)制作,该联盟由UCSC生物分子工程副教授Benedict Paten和生物分子工程助理教授Karen Miga共同领导,现在可以在UCSC基因组浏览器上的组装中心使用。十多名UCSC研究人员和学生是该项目的贡献者,该项目将持续到2024年,届时研究人员计划发布包含350个人基因组信息的最终泛基因组。
“我们通过对不同的人进行抽样并将他们纳入每个人都可以使用的结构中,从而在参考文献中引入更多的多样性和公平性,”主要标记论文的资深作者Paten说。“一个基因组不足以代表每个人 - 泛基因组最终将是具有包容性和代表性的东西。
了解基因组变异
每个人的基因组略有不同 - 平均与下一个人相比约0.4% - 了解这些差异可以深入了解他们的健康状况,帮助诊断疾病,预测医疗结果并指导治疗。使用泛基因组参考将提高科学家在未来研究中检测和理解变异的能力。
通常,当科学家和临床医生研究个体的基因组以寻找变异时,他们会将该个体的DNA与标准参考的DNA进行比较,以确定一个或多个碱基对的差异。到目前为止,参考基因组主要由每条人类染色体的单个序列表示,主要来自一个人。但是,这个参考已经有近20年的历史了,并且从根本上受到限制,因为它不能代表人类中存在的遗传变异的财富。这在基因组分析中引入了一个称为参考偏差的问题。
相比之下,新的泛基因组是一个参考,结合了来自不同祖先背景的47个个体的基因组。泛基因组在序列具有相同碱基的区域看起来像线性参考,并扩展以显示存在差异的区域。它同时代表了人类基因组序列的许多不同版本,并为科学家提供了一个更准确的变异比较点,这些变异存在于某些人群中,而其他人群则不存在。
“一个基因组不可能代表我们知道可以在世界各地观察和研究的所有丰富变异,”UCSCHPRC生产中心主任Miga说。“人类泛基因组参考的第一目标是试图扩大参考资源的代表性,使其对研究人类物种更具包容性和更公平,作为参考文献的集合,而不仅仅是一个参考文献。
基因组变异可以是小的,由一个或几个DNA碱基的差异组成,也可以是大的结构变异,被归类为50个碱基对或更大的变异。这些较大的结构性变异可能对健康产生重要影响。到目前为止,由于技术有限和使用单个参考序列的偏见,研究人员一直无法识别人类基因组中存在的70%以上的结构变异。
在泛基因组参考文献中添加的119.90亿个新碱基中,大约有<>万个来自结构变异。结构变异很复杂,可能是序列的反转、插入、缺失或串联重复——重复多次的两个或多个碱基的片段。这些新碱基将帮助研究人员研究基因组中以前没有参考的区域,并有可能在未来的研究中将结构变异与疾病联系起来。
“现在,我们可以映射到更多的结构变体,因此我们正在发现基因组中以前不存在的特征和区域,”Miga说。“这令人兴奋,因为它使我们能够以一种独特的方式看待基因调控,这是我们以前无法研究的,因为这些区域可能会被不恰当地绘制或完全忽略。
与使用标准参考检测相比,使用泛基因组参考进行基因组分析可将结构变异的检测率提高 104%。泛基因组参考还提高了调用小变异的准确性,这些变异只有几个碱基长,大约34%,因为泛基因组中存在的数据量增加。
每个人都携带一组成对的染色体——一组遗传自母亲,另一组遗传自父亲。泛基因组参考中存在的单个基因组包含单倍型解析的信息,这意味着它可以自信地区分两组亲本染色体 - 这是一项重大的科学壮举。拥有这些信息将有助于科学家更好地了解各种基因和疾病是如何遗传的。
这也意味着目前的参考实际上包括94个不同的基因组序列,目标是到700年达到2024个。
创建泛基因组
泛基因组是通过开发先进的计算技术来实现的,该技术将多个基因组序列对齐为一个可用的参考,称为泛基因组图的结构。UCSC计算基因组学实验室的Paten和研究人员帮助领导HPRC开发创建这种泛基因组图结构所需的算法方法。
由于该项目使用的方法,泛基因组参考中的所有基因组都具有极高的质量和准确性,覆盖了每个人类基因组的99%以上,准确率超过99%。
“在线性参考中,我们只有一个序列,每个基因的一个表示,”UCSC生物信息学博士候选人,主要论文的共同第一作者Mobin Asri说。“但我们知道,我们的基因在人群中有不同的变异。使用泛基因组图,我们希望将所有这些变异集中在一个结构中 - 而图是做到这一点的自然方法。
HPRC项目严重依赖长读长和超长读长测序技术来读取生物样本中的DNA。随着最近的进展,这些技术现在可以一次解码数千到数百万个基因组碱基对。然后,长段DNA读数通过专门的算法组装成更完整的基因组序列。理想情况下,每个组装的序列应该代表一条染色体的序列。
长读取包含大约 1% 的时间错误,并且当前的汇编算法并不完美,这可能导致组装的序列在某些位置出错。为了检查和纠正这些错误,已经测序和组装的单个基因组通过多种工具,包括Asri开发的可靠性管道。一旦通过这些工具处理,研究人员就可以确保组件准确和完整。
在通过Asri的管道后,各种基因组通过复杂的算法方法编译成泛基因组图结构。从视觉上看,图基因组允许研究人员将各种参考序列的差异视为其他共享路径中的分歧区域。
构建可访问的资源
泛基因组草案中的所有前47个二倍体基因组都来自参与1000基因组计划(1000G)的个人,这是一项有影响力的努力,从公开同意的样本中创建了一个常见的人类遗传变异目录,并于2015年完成。这些样本的开放同意状态允许任何研究人员访问资源,而没有通常伴随基因组研究的隐私障碍,目的是使尽可能多的人可以访问泛基因组。
“成为一种共同资源是人类泛基因组参考成功的基础,”米加说。“它必须有能力在世界各地对所有研究人员开放,这样我们才能将其用作基础。
HPRC团队专注于外展,以确保泛基因组是一种有用的资源,将在世界各地的诊所中使用。这意味着促进使用泛基因组参考进行研究的研究人员的注释、反馈和输入。
“泛基因组草案是一个重要的原则证明,我们希望这将影响很多人,让他们思考泛基因组以及它可能如何影响他们的工作,”帕滕说。“展望未来,我们看到了很多与其他团体的接触——需要很多不同的人来建立一些将成为大型社区资源的东西。
除了关注可访问性外,HPRC项目还有一个专门的道德团队,专注于该项目的社会和法律影响。他们正在努力预测具有挑战性的问题,并帮助指导知情同意,优先考虑不同样本的研究,探索与临床采用有关的可能监管问题,并与国际和土著社区合作,将他们的基因组序列纳入这些更广泛的努力。
继续传承和未来工作
人类泛基因组是加州大学圣克鲁斯分校科学家数十年来努力的延续,以了解人类生命背后的生物密码。
2000年,当时是UCSC研究生,现在是基因组学研究所的研究科学家和UCSC基因组浏览器主任的Jim Kent编写了组装人类基因组第一份工作草案的代码。UCSC科学家发布了它,任何想要使用它的人都可以访问。从那时起,UCSC一直处于基因组学研究的最前沿。
2022年<>月,UCSC的Karen Miga共同领导了端粒到端粒联盟,组装了第一个完整的人类基因组测序,填补了科学家长期以来一直没有的缺失的复杂参考区域。
“自2000年以来,我们对一个基因组进行了一系列越来越准确的表示,”UCSC基因组学研究所科学主任David Haussler说,他领导UCSC团队进行原始人类基因组计划,并为泛基因组计划提供建议。“但无论你多么准确地代表一个基因组,这都不会代表全人类。现在是一个转折点:不再是单一标准人类基因组的基因组学,而是每个人的基因组学。
研究人员正在朝着到2024年完成整个泛基因组的目标取得进展。该团队正在招募新的个体来代表一些未包含在1000基因组计划中的人,特别是中东和非洲血统的人。Miga作为UCSC数据生产中心的主任,将带头推进这些工作。
除了完成最终的泛基因组参考外,研究人员还致力于组建一个国际人类泛基因组项目,与世界各地的研究人员建立伙伴关系。这些伙伴关系将包括双向技能和知识交流,旨在将创建高质量参考基因组所需的技能和技术带到全世界的研究人员手中,以便他们能够开展自己的研究。
免责声明:本文由用户上传,与本网站立场无关。财经信息仅供读者参考,并不构成投资建议。投资者据此操作,风险自担。 如有侵权请联系删除!
-
安徽淮南地区的长安汽车经销商近期对2025款启源Q05车型的市场策略进行了调整,以进一步吸引消费者。作为长安汽...浏览全文>>
-
近年来,豪华SUV市场持续升温,而作为大众旗下的高端品牌,途锐凭借其卓越的性能与豪华配置,一直深受消费者的...浏览全文>>
-
在新能源汽车市场中,一汽-大众ID 4 CROZZ凭借其出色的性价比和丰富的配置吸引了众多消费者的关注。作为一款...浏览全文>>
-
近年来,随着国内汽车市场的快速发展,越来越多的消费者开始关注性价比高的小型车。QQ多米作为一款备受关注的...浏览全文>>
-
在当今的汽车市场中,选择一款高性价比的车型是许多消费者的重要考量。对于追求品质与经济平衡的购车者来说,...浏览全文>>
-
阜阳地区消费者对上汽大众途岳的关注度一直很高,尤其是2025款途岳的上市更是引发了广泛关注。作为一款紧凑型S...浏览全文>>
-
天津滨海长安猎手K50作为一款备受关注的车型,在市场上拥有较高的关注度。这款车型以其出色的性能和合理的价格...浏览全文>>
-
近年来,随着新能源汽车的普及,插电混动车型因其兼顾燃油经济性和驾驶性能的特点,受到了越来越多消费者的青...浏览全文>>
-
途观X作为上汽大众旗下的高端轿跑SUV车型,凭借其时尚动感的外观设计和丰富的科技配置,在市场上一直备受关注...浏览全文>>
-
在选择SUV车型时,上汽大众途昂凭借其宽敞的空间和强大的性能成为不少消费者的首选。对于蚌埠地区的消费者来说...浏览全文>>
- 天津滨海长安猎手K50多少钱 2025款落地价,最低售价18.29万起,赶紧行动
- 天津滨海ID.4 CROZZ 2025新款价格,买车省钱秘籍
- 安徽淮南途昂X多少钱?性价比超高的选车秘籍
- 福特领睿试驾,开启完美驾驭之旅
- 比亚迪海豹05DM-i试驾预约流程
- 凯迪拉克CT5预约试驾,从预约到试驾的完美旅程
- 滁州途观X落地价,各配置车型售价全知晓
- 极狐 阿尔法T6试驾,开启完美驾驭之旅
- 长安欧尚520试驾操作指南
- QQ多米试驾需要注意什么
- 长安Lumin试驾,感受豪华与科技的完美融合
- 安庆长安猎手K50最新价格2025款与配置的完美平衡
- 池州迈腾GTE最新价格2022款全解,买车必看的省钱秘籍
- 安徽亳州ID.6 X多少钱?购车全攻略来袭
- 五菱雪宝试驾,开启豪华驾驶之旅
- 马自达EZ-6预约试驾,新手必看的详细流程
- 安徽亳州T-ROC探歌落地价实惠,配置丰富,不容错过
- 滁州高尔夫GTI价格大揭秘,买车前必看
- 东莞威然落地价,最低售价22.99万起优惠不等人
- 试驾王牌M7,体验豪华驾乘的乐趣